LÄHDE: Juleen R. Zierath,Anna Krook: Insulinresistens I skelettmuskeln på molekylär nivå
T2DM, Sivuilta 37-46
TAUSTA
Tyypin 2 diabetes mellitus on koko
kehoa kohtaava tauti ja antaa oireita, kun useiden elinten
yhteistoiminta on alkanut huonontua. Otsikon kirjanen valaisee tätä
monessa eri kappaleessa. Sekä perintö- että ympäristötekijät
vaikuttavat. Diagnoosi tehdään kohonneesta verensokerista.
Glukoosin insuliinista riippuvan solunoton primaarinen kohde elin on
luustolihas, koska se ottaa noin 80% glukoosista vasteena
insuliinille. Luustolihasta on noin puolet kehon painosta. Sen takia
luustolihaksen aineenvaihdunnan muutokset vaikuttavat koko kehon
aineenvaihdunnalliseen tasapainoon.
Tyypin 2 diabetesta potevalla on
insuliiniin vastaava glukoosin soluunotto huomattavasti alentunut ja
se on tyypillinen löydös selvässä T2DM:ssa ja
glukoositoleranssin alentumassa, mutta osoitettavissa myös
terveistä, joiden 1. asteen sukulaisella on T2DM. Kirjassa
selitetään, miten T2DM ilmenee solutasolla.
1. Miksi lihas ai anna asianmukaista vastetta insuliiniin?
Viimeiset
kaksikymmentä vuotta on
intensiivisesti tutkittu
niitä luustolihassolun
molekyylejä,
jotka välittävät
insuliinisignaalia glukoosin soluunoton lisäämiseksi.
Glukoosimolekyyli ei pysty diffundoitumaan lihassoluun itsestään,
vaan se tarvitsee kuljetusproteiineja mennäkseen
plasmakalvon läpi. Tämän
takia kaikissa kehon soluissa ilmenee erilaisia glukoosin kuljettajia
mahdollistamassa glukoosin pääsyn
soluun.
GLUT4-
niminen glukoosin kuljettaja ilmenee lihaksessa ja rasvakudoksessa.
Perustilassa GLUT4 on todettavissa solun sisällä.
Vasteena insuliinisignaalille GLUT4 siirtyy
plasmakalvoon ja mahdollistaa glukoosin pääsyn
soluun. T2DM-taudissa tämä
vaihe on viallinen
lihassolussa. Kyky rekrytoida GLUT4 plasmakalvoon on T2DM-taudissa
alentunut. Euglykemisen hyperinsulinemian insuliiniresistenssi
selittyykin siitä suurelta
osaltaan. Glukoosin soluunotossa on 40% n alenema T2DM-tautia
potevilla verrattuna terveisiin.
2. Miten insuliini lisää lihassolun glukoosinottoa?
Kohonneeseen verensokeriin vasteena
haimasolut vapauttavat vereen insuliinia. Insuliinia sitoutuu sitten
spesifiseen reseptoriin, joita lihassolun kalvossa on. Sitoutuminen
johtaa insuliinireseptorissa tapahtuvaan rakenteelliseen muutokseen
solukalvon sisäpinnalla
sijaitsevassa osassa ja samalla siinä käynnistyy
reseptorin tyrosiinikinaasientsyymiaktiivisuus. Seuraa
tyrosiinien fosforyloituminen itse reseptorissa ja myös
muissa substraateissa. Entsyymikohteista olennaisimpiin kuuluu IRS-1,
insuliinireseptorisubstraatti-1,
joka saatuaan tyrosiinifosforylaation panee alkuun signaalikaskadin,
mikä johtaa lopulta GLUT4-
molekyylejä kantavien
rakkuloiden siirtymiseen ja sulautumiseen
solukalvoon.
On osoitettu diabeettisella
lihaksella olevan huontunut kyky fosforyloida IRS-1
molekyylin tyrosiineja. Siitä seuraa
kaikkien alavirtasignaalien vaimentuminen: PI3K, PDK1, AKT/PKB,
TBC1D4.
T2DM -potilailta
on osoitettu P13K-
aktivoitumisen alassäätyminen.
Tämä entsyymi on
fosfatidyyli-inositoli -3-
kinaasi.
Muidenkin olennaisten
signaalimolekyylien fosforyloituminen on alentunutta
diabeteslihaksessa, kuten AKT
-fosforylaatio. Akt on sama
kuin PKB.
TBC1D4 fosforyloituminen on myös alentunut.
TBC1D4 fosforyloituminen on myös alentunut.
Grönlannissa
on suhteellisen tavallinen löytö TBC1D4
mutaatio. Se on suoraan linkkiytynyt lihaksen alentuneeseen
insuliiniherkkyyteen ja lisääntyneeseen
T2DM- riskiin, mikä
korostaa tämän
extrasellulaarisen signalointitien merkitystä.
Koska AKT/PKB
säätelee useita
aineenvaihdunnallisia
prosesseja, alentunut aktivoituminen tuo muassaan muutoksen
glykogeenin varastoitumiseen ja proteiinien rakentumiseen.
Tarkkaa tietoa ei ole vielä
siitä, miksi
insuliinireseptori ei pysty T2DM potilaiden lihaksessa fosforyloimaan
IRS1 molekyylin tyrosiineja. Toisaalta on voitu osoittaa, että
vahva seriinien fosforyloituma
IRS molekyylissä estää
insuliinireseptoria pääsemstä
käsiksi
IRS molekyylissä
sijaitseviin tyrosiineihin.
Useat kinaasit fosforyloivat IRS-
molekyylin spesifisesti seriinikohtiin. Niihin kuuluu PKC
eli proteiinikinaasi C, jonka lipidit
aktivoivat. Samoin IKKB,
jota aktivoivat
tulehdussignaalit ja myös
PKC. Osaltaan siten selittyisikin, miksi juuri lipidit ja tulehdukset
johtavat insuliiniresistenssiin. Todennäköistä
on kuitenkin, että IRS-
molekyylin lisääntyneen
seriinifosforyloitumisen taustalla on useita
erilaisia tekijöitä ja se
voi olla erilaista eri T2DM alaryhmienkin kesken.
Fosfataasienkin
aktiivisuus seriinikinaasien kohonneen aktiivisuuden ohella
vaikuttaa intrasellulaaristen signaalien välittymisiin.
Fosfataasit ovt entsyymeitä,
jotka poistavat fosfaatteja
seriinikohdista ja tyrosiinikohdista. Kuitenkin fosfataasien säätymistä
T2DM henkilöillä
on tutkittu vähemmässä
määrin.
Mitä
geenieroja on T2DM potilaiden lihaksen
ja normaalin glukoosinsiedon
omaavien lihaksen kesken?
Tutkimuksissa
on käytetty microarray analyysiä ja on
tunnistettu T2DM -potilailla alasäätymistä
geeneissä, jotka
kontrolloivat oksidatiivista fosforylaatiota ja mitokondriatoimintaa.
Artikkelin kirjoittajat ovat yllä mainitulla
menetelmällä todenneet DGKdelta-entsyymin olevan alassäätyneentä
T2DM-henkilöiden lihaksessa. Tämä entsyymi on
diasyyliglyserolikinaasi delta (DAG kinaasi
delta).
Nämä DAG- kinaasit ovat solun rasvametaboliaa sääteleviä entsyymeitä. Solun sisällä ne kontrolloivat kahden tärkeän signaalivälittäjän keskeistä tasapainoa (DAG, IP3). DGK-delta katalysoi DAG:in hajoamista.
DAG sinänsä aktivoi PKC-entsyymiä.
Samanaikaisesti DAG- muodostumisen kanssa solun sisällä muodostuu IP3- signaalivälittäjää, liukoista inositoli-3-fosfaattia, ja se on tärkeää rakkuloiden siirtymiselle ja lisää solunsisäistä Ca++ pitoisuutta.. (Nämä molemmat signaalivälittäjät ovat alkuisin PIP2-molekyylin hajoamisesta).
Jos fosfoinositideistä (PI-järjestelmästä lipositoleista) purkautunut DAG ei saa DAG-kinaasiaan, ja muutu fosfatidaatiksi (PA), joka voi taas (CTP- energian avulla) palata (salvage)kiertoon lipidipuolella: PIP, PIP2, PIP3), jota insuliinisignalointi hyödyntää, vaan joutuu PKC- entsyymin metabolia-alueelle, huonontuu insuliinin stimuloima glukoosin soluunotto. Niillä T2DM-potilailla, joilla paastosokerit olivat korkeimpia, lihaksen DGKdelta entsyymin pitoisuudet olivat matalinta tasoa. Tämä voi tarkoittaa sitä, että, useimpien diabeetikoiden alentuneet DGK-delta-tasot ovat seurausta kohonneista verensokeriarvoista, jotka taas puolestaan edelleen huonontavat insuliiniherkkyyttä.
Nämä DAG- kinaasit ovat solun rasvametaboliaa sääteleviä entsyymeitä. Solun sisällä ne kontrolloivat kahden tärkeän signaalivälittäjän keskeistä tasapainoa (DAG, IP3). DGK-delta katalysoi DAG:in hajoamista.
DAG sinänsä aktivoi PKC-entsyymiä.
Samanaikaisesti DAG- muodostumisen kanssa solun sisällä muodostuu IP3- signaalivälittäjää, liukoista inositoli-3-fosfaattia, ja se on tärkeää rakkuloiden siirtymiselle ja lisää solunsisäistä Ca++ pitoisuutta.. (Nämä molemmat signaalivälittäjät ovat alkuisin PIP2-molekyylin hajoamisesta).
Jos fosfoinositideistä (PI-järjestelmästä lipositoleista) purkautunut DAG ei saa DAG-kinaasiaan, ja muutu fosfatidaatiksi (PA), joka voi taas (CTP- energian avulla) palata (salvage)kiertoon lipidipuolella: PIP, PIP2, PIP3), jota insuliinisignalointi hyödyntää, vaan joutuu PKC- entsyymin metabolia-alueelle, huonontuu insuliinin stimuloima glukoosin soluunotto. Niillä T2DM-potilailla, joilla paastosokerit olivat korkeimpia, lihaksen DGKdelta entsyymin pitoisuudet olivat matalinta tasoa. Tämä voi tarkoittaa sitä, että, useimpien diabeetikoiden alentuneet DGK-delta-tasot ovat seurausta kohonneista verensokeriarvoista, jotka taas puolestaan edelleen huonontavat insuliiniherkkyyttä.
Tältä kannalta on kiinnostavaa,
että kohonneen verensokeritason hoitaminen
normalisoi lihaksen DGK-delta- entsyymin tason.
Myös fyysinen aktiivisuus lisää
lihaksen DGK-delta-entsyymimääriä, mikä osoittaa, kuinka eri miljöötekijät voivat suoraan vaikuttaa
lihaksen insuliinisignalointiin.
- (VIITE: antaa käsitystä DGKdelta entsyymiryhmästä. Esimerkki on T-solualueelta)
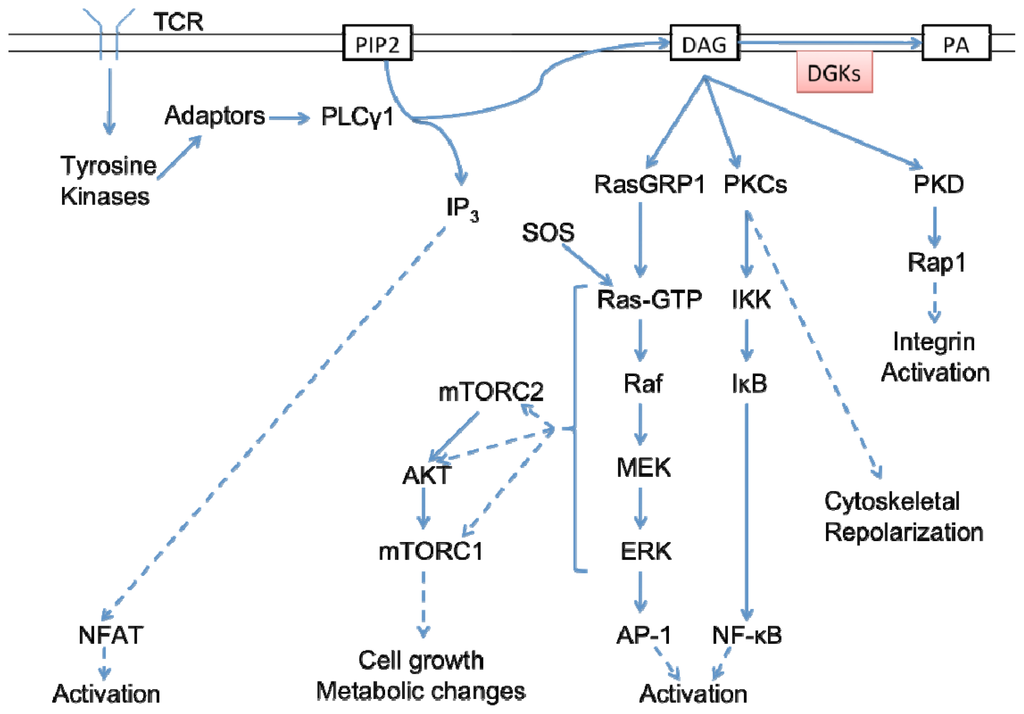
- Receptor-mediated signaling is critical for immune cell development and function. Engagement of the T cell receptor (TCR) on T cells activates signaling cascades that lead to their differentiation, proliferation, and elaboration of cytokines, which are required for optimal immunity. After engagement of the TCR by peptides displayed on antigen presenting cells (APCs), signaling is amplified by the activation of phospholipase C γ1 (PLCγ1), which cleaves phosphatidylinositol-4,5-biphosphate (PIP2) to form the second messengers diacylglycerol (DAG) and inositol triphosphate (IP3). Synthesis of DAG is crucial for activation of diverse downstream signaling cascades, including the Ras, NF-κB, and AKT pathways. DAG levels must therefore be finely tuned not only through controlled production but also by its metabolism.
- Diacylglycerol kinases (DGKs) are a diverse family of enzymes that phosphorylate DAG to form phosphatidic acid (PA), thereby terminating DAG signaling and initiating additional signaling events through the synthesis of bioactive PA.
- The critical role of DGKs in controlling DAG in T cells has become evident over the last decade through the use of both cell line and in vivo models.
- The regulation of DGKs themselves, however, has begun to be understood only recently. This review summarizes our understanding of the role of DGKs in T cells and describes new advances in deciphering the means by which DGKs are regulated).
{kind=link}
Mutta
DAG sinänsä aktivoi PKC-entsyymiä, joka
sijaitsee solun sisällä.
Jo kauan on tiedetty,että signaalista samanaikaisesti
DAG- muodostumisen kanssa solun sisälle
muodostuu IP3- signaalivälittäjää, liukoista
inositoli-3-fosfaattia, ja se on tärkeää rakkuloiden siirtymiselle
ja lisää solunsisäistä Ca++ pitoisuutta. (Nämä molemmat
signaalivälittäjät ovat alkuisin PIP2-molekyylin hajoamisesta -
(DAG on lipidinen ja sisältää tavallisesti
steariinihapon ja arakidonihapon ) ja
IP3 on vesiliukoinen,inositolirenkaassa kolme fosfaattia, asemissa 1,4,5); S e myös toimii ATP-energia-alueella kuten sokerirakenteet).
Jos fosfoinositideistä (PI-
molekyyleistä) purkautunut DAG ei saa heti
DAG-kinaasiaan,
(eli: päätä signalointiaan ja
muutu fosfatidaatiksi (PA), joka voi taas CTP-
energian avulla palata (salvage)
kiertoon PIP, PIP2), jota
insuliinisignalointi hyödyntää,
vaan jos se
joutuu PKC- entsyymin havaitsemaksi
ja sen metabolia-alueelle, huonontuu insuliinin
stimuloima glukoosin soluunotto.
Niillä T2DM-potilailla, joilla
paastosokerit olivat korkeimpia, lihaksen DGKdelta -entsyymin
pitoisuudet olivat matalinta tasoa. Tämä voi
tarkoittaa sitä, että, useimpien diabeetikoiden alentuneet
DGK-delta-tasot ovat seurausta kohonneista verensokeriarvoista, jotka
taas puolestaan edelleen huonontavat insuliiniherkkyyttä.
Tältä kannalta on kiinnostavaa,
että kohonneen verensokeritason hoitaminen
normalisoi lihaksen DGK-delta- entsyymin tason.
Myös fyysinen aktiivisuus lisää
lihaksen DGK-delta-entsyymimääriä,
mikä osoittaa, kuinka eri miljöötekijät voivat suoraan vaikuttaa
lihaksen insuliinisignalointiin.
3. Mikä vaikuttaa lihaksen insuliiniherkkyyteen?
Useat tekijät
vaikuttavat siihen, miten hyvin lihas reagoi insuliiniin. Esimerkiksi
jos infusoidaan suoneen rasvaemulsiota, vähenee insuliiniherkkyys
nopeasti. Insuliiniherkkyyden muutokseen on liitetty useita erilaisia
veressä kiertäviä tekijöitä
ja niistä moni on peräisin rasvakudoksesta. Esimerkkejä ovat
leptiini, adiponektiini ja tulehdusmerkitsijät. Lihassupistuksella
ja fyysisellä aktiivisuudella on dokumentoitu hyvä vaikutus
lihaksen insuliiniherkkyyteen.
Lihastyöllä on välitön
vaikutus, koska glukoosinotto lihassoluun
lisääntyy silloin insuliinista riippumattakin. Se johtuu
GLUT4-molekyylin translokaatio. Lihassupistus johtaa moniin
solumuutoksiin. Esimerkiksi venytykselle herkät reseptorit
aktivoituvat. Solun sisäinen pH muuttuu. Jonisoitunutta kalsiumia
ja muita metaboliitteja vapautuu. Samalla myös ATP/AMP-suhde
muuttuu.
Kalsiumin (Ca++) vapautuminen
johtaa monen eri signaaliketjun
aktivoitumiseen ja niistä moni sinänsä voi vaikuttaa lihaksen
aineenvaihduntaan ja
insuliiniherkkyyteen.
AMPK
(AMP-molekyylin, adenosiinimonofosfaatin, aktivoima
proteiinikinaasi) tiedetään
tärkeäksi signaalimolekyyliksi ja sen aktivoituminen voi johtaa
GLUT4-molekyyliä sisältävien rakkuloiden siirtymiseen lihassolun
plasmakalvoon. AMPK aktivoituu, kun ATP-energiapakettien (
trifosfaattien) määrä laskee suhteessa AMP- määrään
(monofosfaattien määrään), toisin sanoen ATP/AMP suhteen
laskiessa. Tehtäessä lihastyötä
kulutetaan ATP:tä ja sen seurauksena ATP-pitoisuudet laskevat ja
AMP-pitoisuudet nousevat ja AMPK-entsyymi aktivoituu. Myös muut
olosuhteet voivat aktivoida AMPK-entsyymin,
kun ATP-pitoisuudet jostain syystä laskevat: esimerkiksi
ravinnonoton laskiessa tai solustressin vallitessa.
Kun AMPK on
aktiivina, lisääntyy lihaksessa glukoosin
soluun otto insuliinista riippumattomalla tavalla. Samanaikaisesti
lisääntyy rasvahappojen poltto energiaksi , kun solu koettaa
palauttaaa
energiatasapainoaan. Näin ollen AMPK:n
aktivoiminen on eräs mahdollisuus vaikuttaa
lihaksen glukoosin oton lisääntymistä ja samalla voidaan kiertää
alassäätyneen insuliinisignaloinnin ongelmistoa.
- VIITE: BAIB, tymiinin luonnollinen metaboliitti, jota muodostuu lihastyössä.
Pitkäaikainen treenaus ja
fyysinen aktiviteetti johtaa enemmän tai
vähemmän välittömiin AMPK-entsyymin aktivoitumisiin ja
treenatussa lihaksessa myös geeni-ilmenemän muutokseen ja yhdessä
nämä seikat johtavat lihaksen parempaan insuliiniherkkyyteen. Muun
muassa treenausvasteena lisääntyy GLUT4 proteiini. Myös
mitokondrioitten lukumäärä ja toiminta vaikuttuvat kohentuneeseen
suuntaan.
On vielä paljon tieteellisen
tutkimuksen aihetta siinäkin, miten nämä liikunnan hyvät
aineenvaihdunnalliset vaikutukset välittyvät molekyylitasolla,
sillä lihassupistuksestahan aktivoituu tai vaimenee moni eri
signaaliketju, ja aktivoitumisaste riippuu lihastyön tavasta,
intensiteetistä ja kestosta.
4. Mitokondrioitten aineenvaihdunnallinen jähmeys
Mitokondria on solunsisäinen
organelli ja se hajoittaa erilaisia energia-aineita. Mitokondriat
voivat tehdä vaihteen eri
pääenergia-aineiden käytön kesken. Pääenergia-aineet ovat
hiilihydraatit (kuten glukoosi), rasvat ( eri
rasvahapot) ja proteiinit (aminohapot).
Insuliinin stimuloimassa tilanteessa mitokondria käyttää
glukoosista johtuvia orgaanisia hiiliketjumuotoja. Väliaikoina
mitokondriat käyttävät ennen kaikkea
rasvahapoista peräisin olevia hiiliketjuja, kun
ne generoivat
ATP:tä.
Tämä kyky pystyä tekemään
vaihde eri energiapäälajien substraatin kesken on
aineenvaihdunnallista joustavuutta (fleksibiliteettiä). Tämä kyky
on alentunut T2DM:ssa. Nykyisissä tutkimuksissa keskitytään tämän
mitokondriaalisen metabolisen inflexibiliteetin
(aineenvaihdunnallisen jähmeyden) taustasyitten ymmärtämiseen.
5. Rasvahapot ja metyloitumisaste
Erilaisiin proteiineihin voi
linkkiytyä metyyliryhmiä, myös DNA:n cytosiinikohtiin, jolloin
kyse on epigeneettisestä modifioitumisesta. Usein DNA:n
metyloituminen vaikuttaa siihen, miten DNA on erilaisten
proteiinitekijöiden kuten esim. transskriptiotekijöitten
saatavilla. Sen takia metyloitumisaste voi säädellä, miten
DNA-sekvenssit ovat aktiiveja ja mitkä geenit ilmenevät.
T2DM- henkilöiltä voidaan
analysoida lihaksen promoottori-DNA:n metyloitumisaste ja
vertailla tätä terveisiin kotnrollihenkilöihin. Tämän
artikkegeenin ilmenemä) lin kirjoittajat havaitsivat
metyloitumisasteita tutkiessaan T2DM-potilailla noin 800 geenissä
merkitseviä eroja. Näistä geeneistä mainitaan esimerkkinä
PGC1alfa geeni. T2DM-henkilöiden lihaksessa oli tämän geenin
promoottorin DNA-sekvenssi (= se osa DNA:ta, josta säätyy geenin
ilmenemä) oli korkea-asteisemmin metyloitunut kuin terveillä .
Samaa havaittiin myös niiltä henkilöiltä, jotka potivat
vaikea-asteista lihavuutta. Mielenkiintoista oli sekin, että
ylipainoa vähentävän kirurgisen operaation jälkeen
metyloitumisaste palautui samaan kuin insuliiniherkillä
normaalipainoisilla henkilöillä.
Myös T2DM- henkilöiden
beetasoluissa on havaittu saman geenin PGC1alfa promoottorin
metylaatioasteen muuntuneen. Tutkijat tekivät myös sen havainnon,
että beetasolun PGC1alfa-geenin promoottorin metyloitumisaste
korreloi lihaksen ilmentämään PGC1alfa-määrään
käänteisesti: Jos PGC1alfa:n promoottorin metyloitumisaste oli
beetasolussa korkea, PGC1alfan ilmenemä lihaksessa oli matala.
Niinpä T2DM-henkilöillä voi
PGC1alfa-geenin vahvassa metyloitumisasteessa olla syy lihaksen
vähempään PGC1alfa-ilmenemään.
Koeputkiviljelmistä tehtiin
mielenkiintoinen havainto: Viljeltyjen lihassolujen PGC1alfa-geenin
promoottorin metyloitumisaste nousi suorana vasteena väliaineen
rasvahappojen aiheuttamaan altistukseen, mikä osoittaa, että
lihassolua ympäröivä miljöö voi vaikuttaa metyloitumisasteeseen.
Entsyymi DNMT3B.
Rasvahappojen aiheuttama
PGC1alfa-promoottorin metylaatio riippui eräästä spesifisestä
metyylitransferaasista DNMT3B siten, että solut joista tämä
entsyymi puuttui, olivat suojattuja rasvahappojen
metyloitumisastetta kohottavalta vaikutukselta ja PGC1alfaan
kohdistuvalta vaikutukselta.
On vielä epäselvä asia, millä
tavalla rasvahapot ovat ja ovatko ne ylipäätänsä se tekijä,
joka säätelee PGC1alfageenin promoottorin metyloitumisasteen.
Aiemmista tutkimuksista tiedetään kuitenkin, että elintavan
muutokset painonlaskuineen ja lisättyine fyysisine aktiivisuuksineen
on ylipainoisilla liittynyt mitokondrioitten lukumäärän ja koon
lisääntymiseen. Nämä löydöt osoittaisivat, että
insuliiniresistenssin vallitessakin lihas on säilyttänyt tallella
sekä kykynsä vastata fyysiseen aktiviteettiin että
mitokondriaalisen plastisuutensa.
Mitokondrioitten koon ja lukumäärän
lisääntymiset liittyivät insuliiniherkkyyden kohentumisiin.
Rajoitetussa määrin käytetään
nykyisin eräissä tuumoritaudeissa lääkkeitä, jotka vaikuttavat
metyloitumispotentiaaliin. Edellämainitut tulokset laajentanevat
indikaatioaluetta ehkä metabolistenkin tautien hoidon alueelle.
6. Perimä, ympäristö ja epigenetiikka
Tuleva tutkimus selventänee, missä
määrin epigeneettiset muutokset antavat osuutta perimän ja
ympäristön yhteisvaikutuksiin T2DM:n kehittymisessä. On myös
tärkeä oivaltaa, jos ja miten ympäristötekijät kuten dieetti ja
fyysinen aktiviteetti vaikuttavat eri geenien metyloitumisstatukseen
ja pidemmällä ajalla geenifunktioon. Tärkeää on myös ymmärtää
hyödyntää tietoa metabolisten tautien hoitoon ja ennaltaehkäisyyn.
LÄHDE:
Medicinsk kunskap från
Läkartidningen. DIABETES TYP 2.
Claes -Göran Östenson (red) .
Läkartidningen Förlag AB Sidorna 37- 46.
Juleen R. Zierath,Anna Krook:
Insulinresistens I skelettmuskeln på molekylär nivå
Suomennos L Bright 9.1.2017
Inga kommentarer:
Skicka en kommentar